Was ist ALS?
Die Amyotrophe Lateralsklerose (ALS) ist eine schwere Erkrankung des motorischen Nervensystems. Bei der ALS kommt es zu einer Degeneration von motorischen Nervenzellen (auch Motoneurone genannt), die für die willkürliche Steuerung der Muskulatur verantwortlich sind. In der Folge der ALS entstehen fortschreitende Lähmungen (Paresen) oder eine unkontrollierte Muskelanspannung (Spastik) der Willkürmotorik. Für die Betroffenen stellt sich der Nervenzellverlust als Fortschreitende Einschränkung der Mobilität der Hände, Arme, Beine sowie des Rumpfes und der Zunge dar. Im fortgeschrittenen Krankheitsverlauf kann es zur vollständigen Lähmung der Skelettmuskulatur kommen. Die ALS zählt daher zu einer der schwersten Erkrankungen des Menschen. Sie ist nicht heilbar, durch eine Behandlung können ihre Symptome jedoch gelindert werden.
In Deutschland leben 6.000 bis 8.000 Menschen mit ALS. Die Erkrankung wird jährlich bei etwa 2.000 Patientinnen und Patienten neu diagnostiziert. Die meisten von ihnen sind zwischen 50 und 70 Jahre alt. Das mittlere Alter bei Erkrankungsbeginn liegt bei 60 Lebensjahren. 10 % der Menschen mit ALS erkranken vor dem 40. Lebensjahr. Die jüngsten Patientinnen und Patienten sind zwischen 20 und 30 Jahre alt. Männer erkranken etwas häufiger als Frauen.
FAQ
-
Was bedeutet der Begriff „Amyotrophe Lateralsklerose”?
Die Abkürzung ALS steht für den medizinischen Begriff „Amyotrophe Lateralsklerose“. Es handelt sich um den medizinischen Namen, den der Erstbeschreiber der ALS für diese Erkrankung im Jahre 1874 vorgeschlagen hat.
Dieser medizinhistorische Begriff beschreibt Grundelemente der Erkrankung. „Amyotroph“ lässt sich mit der Formulierung „ohne Muskeln“ übersetzen. Das Wort „Lateralsklerose“ steht für eine „seitliche Verkalkung“. Diese Formulierung zielt auf den Abbau des Seitenstranges im Rückenmark, der die zentrale motorische Nervenbahn im Rückenmark verkörpert und bei der ALS degeneriert.
Eine freie Übersetzung des Begriffes der Amyotrophen Lateralsklerose bedeutet „Muskelschwund durch einen Abbau der Seitenstränge im Rückenmark“. Dieser Begriff ist ausschließlich historisch zu verstehen, da in der Begrifflichkeit nur die Veränderungen auf Rückenmarksebene beschrieben werden und auch der Lähmungscharakter im Wort nicht beschrieben wird.
Der ALS-Begriff ist damit inhaltlich nicht „korrekt“, aber ein weltweit verbindlicher Name der zugrunde liegenden Erkrankung. Der weite Gebrauch eines historischen Krankheitsbegriffes ist nicht nur für die ALS typisch, sondern betrifft die gesamte Medizin. Beispiele für weitgenutzte historische Begriffe sind „Multiple Sklerose“, „Krebs“, „Arteriosklerose“, „Schlaganfall“ und zahlreiche Bezeichnungen anderer schwerer Erkrankungen.
-
Was bedeutet „Motoneuron-Erkrankung”?
Der Begriff „Motoneuron-Erkrankung“ lässt sich als „Erkrankung der motorischen Nervenzellen“ übersetzen. „Moto“ steht für das Wort „motorisch“, während das „Neuron“ der medizinische Begriff für „Nervenzelle“ darstellt. Motoneuron-Erkrankungen sind damit die Gesamtheit aller Erkrankungen, bei denen motorische Nervenzellen abgebaut werden.
Bei Motoneuronen-Erkrankungen können die folgenden Symptome auftreten: unvollständige Lähmungen („Parese“ genannt), vollständige Lähmungen („Plegie“ oder „Paralyse“ genannt), Muskelschwund („Myatrophie“ genannt) oder eine unkontrollierte Muskelanspannung, die sich als Muskelsteifigkeit darstellt („Spastik“ genannt).
Die ALS ist die häufigste Motoneuron-Erkrankung. Neben der ALS gehören auch andere Erkrankungen dazu, die diagnostische und prognostische Unterschiede zur ALS-Erkrankung aufweisen. Zur Gruppe der Motoneuron-Erkrankungen, die keine ALS verkörpern, gehören die Spinale Muskelatrophie (SMA), die Spinobulbäre Muskelatrophie (SBMA; auch Kennedy-Erkrankung genannt) sowie die Spastische Spinalparalyse (SSP), die unter bestimmten Umständen auch als Hereditäre Spastische Paraparese (HSP) bezeichnet wird.
Die Symptomverteilung, der Schweregrad und die Dynamik der Symptomentwicklung zwischen diesen Erkrankungen sind sehr unterschiedlich. Für einen Spezialisten oder eine Spezialistin ist die Unterscheidung zwischen diesen Diagnosen möglich. In bestimmten Kliniken wird der Begriff der „Motoneuron-Erkrankung“ auch synonym für die ALS benutzt, da sie die häufigste Motoneuron-Erkrankung des Erwachsenenalters darstellt. In Großbritannien und britisch-geprägten Gesundheitssystemen wird der Begriff Motoneuron-Erkrankung (Motor Neuron Disease; MND) anstelle des Wortes der ALS verwendet.
-
Seit wann ist die ALS bekannt?
Die ALS wurde erstmalig im Jahr 1874 vom französischen Neurologen Jean-Martin Charcot am Pariser Universitätskrankenhaus Hôpital de la Salpêtrière beschrieben.
Er bezeichnete die Erkrankung als „Amyotrophe Lateralsklerose“. Bereits im Jahr 1850 hat der französische Neurologe Aran die progressive Muskelatrophie (PMA) entdeckt. Zum damaligen Zeitpunkt ging man davon aus, dass die PMA und ALS unterschiedliche Erkrankungen seien. Heute ist bekannt, dass die PMA eine spezifische Variante der ALS darstellt. Damit wurde die ALS im weiteren Sinne erstmalig von Aran bereits 1850 charakterisiert.
Die Namensgebung, die bis heute Gültigkeit hat, folgte dann 24 Jahre später durch Charcot.
-
Wer bekommt ALS?
Die ALS ist eine schicksalshafte Erkrankung, für die nach dem heutigen Stand der Medizin keine äußeren Ursachen bekannt sind.
Damit ist die ALS in jedem Fall ohne „Eigenverschulden“ zu betrachten. Bestimmte Erkrankungen sind mit einem Risikoverhalten verbunden (z. B. bestimmte Krebserkrankungen und Herz-Kreislauf-Erkrankungen durch Rauchen, Alkoholgenuss, körperliche Inaktivität usw.). Diese beeinflussbaren Risikofaktoren liegen bei der ALS nicht vor. Daher ist der Begriff der „Schicksalshaftigkeit“ der ALS gerechtfertigt.
Die Mehrheit der Betroffenen war vor der Diagnose einer ALS gesund und ohne wesentliche Vorerkrankungen. Die ALS tritt daher ohne Vorboten auf. Die Mehrheit der Betroffenen erkrankt im Alter zwischen fünfzig und sechzig Lebensjahren. Männer und Frauen sind fast gleichermaßen betroffen: das männliche Geschlecht überwiegt geringgradig (1,5:1). Verschiedene Studien haben versucht, ein bestimmtes Profil von Menschen mit ALS zu identifizieren.
Untersuchungsserien zeigen, dass Menschen mit ALS vor Erkrankungsbeginn schlanker und sportlicher sind als entsprechende Vergleichsgruppen. Weitere Studien haben nachgewiesen, dass Menschen mit ALS – in einer statistischen Betrachtung – einen höheren Bildungsstatus und ein überdurchschnittliches Einkommen aufweisen. Für viele Studienergebnisse zu Persönlichkeitsmerkmalen von ALS-Patientinnen und ‑Patienten liegen auch gegenteilige Untersuchungsergebnisse vor.
Insgesamt lässt sich damit kein „Persönlichkeitsprofil“ für Menschen mit ALS festlegen. Somit kann grundsätzlich jeder Mensch im Verlauf des Lebens an ALS erkranken. Das Risiko für eine ALS ist erhöht, wenn eine familiäre (erbliche) Form der ALS vorliegt.
-
Warum ich?
Die Frage „Warum hat mich die ALS getroffen?“ beschäftigt fast alle Menschen mit ALS. In dieser Frage liegt die Vermutung oder Sorge, dass möglicherweise ein Ereignis in der eigenen Biographie als Krankheitsursache zugrunde liegt, das einem bisher nicht bewusst war.
Die Sorge oder Vermutung ist jedoch medizinisch nicht begründet: für die ALS liegen auch keine Ursachenfaktoren vor, die an einen Lebensstil oder andere biographische Ereignisse gebunden sind. Bestimmte Berufe, Ernährungsgewohnheiten, die Belastung mit Toxinen (Holzschutzmittel, Farben, Lacke und andere Chemikalien), Fremdkörper (Zahnfüllungen, Implantate) oder Infektionen (Borreliose) sind keine Ursachenfaktoren der ALS.
Die Frage „Warum ich?“ lässt sich vereinfacht so beantworten, dass die ALS „zufällig“ entsteht. Hinter diesem „Zufall“ sind bisher unverstandene molekulare Fehler zu vermuten, die zu einer schädlichen Ereignisabfolge auf zellulärer Ebene führen und die Degeneration der motorischen Nervenzellen einleiten.
Zu einem geringeren Teil der ALS-Patientinnen und ‑Patienten sind bereits heute genetische Faktoren (Mutationen in „ALS-Genen“) bekannt, die von vorangehenden Generationen übertragen wurden oder in der eigenen Embryonalentwicklung entstanden sind.
-
Wie viele Menschen in Deutschland leben mit ALS?
In Deutschland sind vermutlich 6.000 bis 8.000 Menschen an ALS erkrankt. Diese Zahl ist eine Annahme, die auf Studienergebnisse zur Häufigkeit der ALS in regionalen oder internationalen Patientenregistern (z. B. dem Schwäbischen oder dem Niederländischen ALS-Register) beruht.
Die exakte Zahl der Betroffenen ist in Deutschland nicht bekannt, da bisher kein bundesweites ALS-Register besteht. Die Studienlage in den bisherigen Registern zeigte eine Häufigkeit von 8 bis 10 Betroffenen pro 100.000 Einwohner. Unter der Annahme, dass die Häufigkeitsverteilung der ALS in der Region Schaben oder den Niederlanden sowie Deutschland weitgehend übereinstimmen, ist bei 80 Millionen Einwohnern in unserem Land von der genannten Zahl von etwa 8.000 Betroffenen auszugehen.
Die Anzahl der Erkrankten pro 100.000 Einwohner wird als ALS-Prävalenz bezeichnet. Davon zu unterscheiden ist die ALS-Inzidenz. Diese Zahl beschreibt die Anzahl der Neuerkrankungen pro Jahr pro 100.000 Einwohner. Bei der ALS ist von etwa 1,5 bis 2 Neuerkrankungen pro 100.000 Einwohner auszugehen. Damit treten in Deutschland 1.200 bis 1.600 Neuerkrankungen pro Jahr auf. Diese Zahl entspricht auch der Anzahl der jährlichen Todesfälle in Folge der ALS in Deutschland.
-
Nach welchen Kriterien ist die ALS eine seltene Erkrankung?
Die ALS erfüllt die formalen Kriterien der Europäischen Union (EU) einer „seltenen Erkrankung“, die mit einer Häufigkeit von weniger als 50 Betroffenen pro 100.000 Einwohner definiert ist.
Die ALS tritt mit einer Häufigkeit von 10 pro 100.000 Einwohnern auf, so dass die genannten Kriterien einer seltenen Erkrankung erfüllt sind. Diese formale Einordnung hat vor allem für die Entwicklung und Zulassung von Medikamenten eine Bedeutung, die in der EU und den USA durch gesetzliche Regelungen gefördert werden. Innerhalb der „seltenen Erkrankungen“ gehört die ALS jedoch zur Gruppe der „häufigen Seltenen“.
-
Warum ist die ALS in Deutschland relativ unbekannt?
Die ALS gehört mit 10 Betroffenen pro 100.000 Einwohner zu den häufigen Erkrankungen innerhalb der Gruppe der „seltenen Erkrankungen“. Trotz der relativen Häufigkeit ist die ALS in Deutschland recht unbekannt.
Die geringe Bekanntheit der ALS ist mit den Besonderheiten der Erkrankung verknüpft: Aufgrund der Einschränkung in Mobilität und Kommunikationsfähigkeit ist es für die Betroffenen eine besondere Herausforderung, Öffentlichkeitsarbeit zu betreiben. Auch die Angehörigen der Betroffenen sind durch die pflegerische und psychosoziale Belastung nur mit großen Einschränkungen in der Lage, eine politische Arbeit zu Gunsten der ALS zu leisten.
Die Anzahl der ALS-Neurologinnen und ‑Neurologen ist in Deutschland ebenfalls sehr klein, so dass die von den ALS-Zentren ausgehende gesellschaftliche Aktivität für eine breite Aufmerksamkeit noch zu gering ist. Erst ab dem Jahr 2002 ist durch die Erkrankung des Künstlers Jörg Immendorff und des Fußball-Bundesligaprofis Krysztof Nowak (VfL Wolfsburg) die ALS in die mediale Öffentlichkeit gerückt. Gemeinsam mit der Charité hat Jörg Immendorff die Öffentlichkeit gesucht, um in Charity-Aktionen, Talkshows und künstlerischen Aktionen auf die ALS aufmerksam zu machen. So unterstützte er im Jahr 2004 die Theaterproduktion „Theater ALS Krankheit“, die von Christoph Schlingensief an der Berliner Volksbühne inszeniert wurde und zu einer weiteren öffentlichen Wahrnehmung der ALS beigetragen hat.
In den weiteren Jahren sind mehrere Fernseh- und Spielfilme entstanden, bei der die ALS ganz im Vordergrund stand: „Sterne leuchten auch am Tag“ (2004), „Hin und weg“ (2014) und „Herbert“ (2016). Auch in internationalen Kino-Produktionen wurde die ALS mehrfach thematisiert: „Die Entdeckung der Unendlichkeit“ (2014) und „Das Glück an meiner Seite“ (2014).
Durch die gesellschaftlichen Aktivitäten von Künstlerinnen und Künstlern, Prominenten und Filmschaffenden ist es gelungen, die Wahrnehmung zugunsten der ALS in Deutschland deutlich zu erhöhen.
Ein wichtiges Ereignis war im Sommer 2014 die internationale „Eiskübel-Aktion“ (englisch: Ice Bucket Challenge). Es handelte sich um die bis dahin größte Spendenaktivität in sozialen Netzwerken, in der – ermöglicht durch „virale“ Effekte des Internets – eine weltweite Spendensumme von mehr als 100 Millionen Euro aufgebracht werden konnte.
Zugunsten der ALS-Ambulanz der Charité haben mehr als 34.000 Spenderinnen und Spender eine Gesamtsumme von 1,6 Millionen Euro gestiftet. Neben den dringend erforderlichen Spenden wurde auch eine hohe gesellschaftliche Aufmerksamkeit in den traditionellen Medien (Fernsehen, Print-Medien) erreicht. Damit gehört die ALS zu den bekanntesten unter den seltenen Erkrankungen.
-
Wie sicher ist die Diagnose einer ALS?
Die Diagnose einer ALS ist bei der Mehrheit der Betroffenen mit hoher Sicherheit zu stellen.
Die Diagnose beruht auf der Anamnese (Arzt-Patienten-Gespräch zur Ermittlung der Krankengeschichte, aus der die genaue Abfolge der Symptome und Beschwerden hervorgeht), dem neurologischen Untersuchungsbefund (körperliche Untersuchung durch einen Facharzt oder eine Fachärztin für Neurologie) und der elektrophysiologischen Diagnostik (Elektroneurographie sowie Elektromyographie). Zusätzlich werden zur Sicherung der Diagnose noch eine Magnetresonanztomographie (MRT) des Kopfes und/oder Rückenmarks durchgeführt. Meist ist auch eine Untersuchung des Nervenwassers (Liquor cerebrospinalis; kurz „Liquor“) erforderlich.
Nicht bei jedem Patienten sind alle Untersuchungen erforderlich, um die Diagnose einer ALS sicher zu stellen. Zumeist ist die Diagnosestellung einer ALS für einen erfahrenen Facharzt oder Fachärztin für Neurologie keine diagnostische Herausforderung.
Nur im Ausnahmefall (weniger als 10 % der Betroffenen) kann die Diagnosestellung komplex und schwierig sein. In der Mehrheit der Krankheitsfälle ist die Diagnose als sicher einzuschätzen. Nur im Ausnahmefall (weniger als 5 %) sind invasive Diagnosemaßnahmen (z. B. eine Muskel- oder Nervenbiopsie) erforderlich. Eine Labordiagnostik zum Nachweis von ALS steht nicht zur Verfügung. Allerdings kann durch die Identifizierung von Molekülen (Biomarker) in Blut, in Nervenwasser (Liquor) oder anderen Körperflüssigkeiten die Diagnose- und Prognosestellung einer ALS weiter erhärtet und ergänzt werden.
Der Biomarker „Neurofilament light chain“ (NF‑L) wird seit 2017 bei bestimmten Situationen zur Diagnosesicherung und in verschiedenen Forschungsprojekten eingesetzt.
Genetische Tests können ebenfalls zur Diagnosestellung beitragen. Sie stehen überwiegend Patientinnen und Patienten mit einer „familiären ALS“ (FALS) zur Verfügung. Eine FALS liegt vor, wenn mehrere Familienmitglieder an einer ALS erkrankt sind und eine erbliche Ursache zugrunde liegt.
In diesem Fall sind genetische Tests verfügbar, der aus einer Blutprobe durchgeführt wird. Bei der Mehrheit der Betroffenen mit einer familiären ALS lässt sich das ursächliche Gen durch die molekulargenetische Analyse nachweisen. Insgesamt lässt sich in der Kombination einer körperlichen Untersuchung und einer elektrophysiologischen Diagnostik (Elektroneurographie, EMG) die Diagnose einer ALS sehr sicher stellen.
-
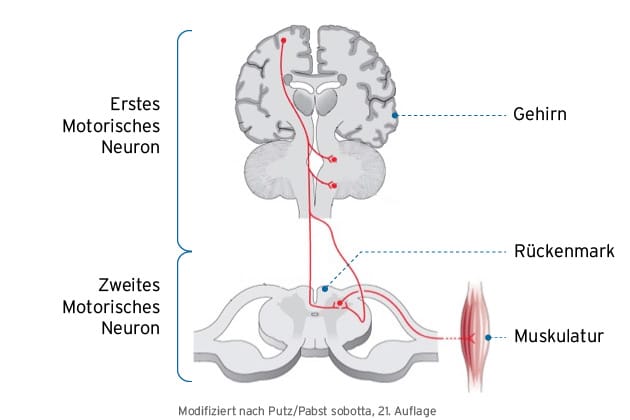
Was bedeutet „erstes Motoneuron” und „zweites Motoneuron”?
„Motoneurone“ (oder auch „motorische Neurone“ genannt) sind Nervenzellen in Gehirn und Rückenmark, die für die Steuerung der Willkürmuskulatur unseres Körpers verantwortlich sind.
Dabei sind grundsätzlich motorische Nervenzellen zu unterscheiden, die sich in der motorischen Hirnrinde („erstes motorisches Neuron“) oder im Rückenmark (oder dem Hirnstamm) befinden („zweites motorisches Neuron“). Die Unterscheidung ist relevant, da bei der ALS beide Nervenzellgruppen – das erste und zweite motorische Neuron – betroffen und deren Schädigung mit unterschiedlichen Symptomen verbunden sind.
Die Schädigung des ersten motorischen Neurons verursacht typischerweise eine unkontrollierte Muskelanspannung (Steifigkeit der Muskulatur), die als Spastik bezeichnet wird.
Die Degeneration des zweiten motorischen Neurons ist wiederum mit Muskelschwund (Myatrophien), Muskelschwäche (Paresen) und mit Muskelzuckungen (Faszikulationen) verbunden.
-
Welche Tests sind notwendig, um eine ALS zu diagnostizieren?
Die ALS wird in erster Linie durch körperliche Zeichen (Muskelschwäche, Muskelatrophie, Reflexsteigerung, Spastik) festgestellt. Neben diesen neurologischen Symptomen werden verschiedene Untersuchungsverfahren eingesetzt, um den klinischen Befund zu bestätigen oder den körperlichen Verdacht auf eine ALS abzuschwächen. Der Umfang von „Tests“, die als Zusatzuntersuchung bezeichnet werden, wird maßgeblich von der Eindeutigkeit der neurologischen Symptome bestimmt. Bei Vorliegen eines eindeutigen, ALS-typischen Untersuchungsbefundes ist die Notwendigkeit von zusatzdiagnostischen Verfahren gering. Die Tests dienen der Bestätigung und Sicherung der klinischen Diagnose. In anderen Situationen kann die klinische Diagnose noch unbestimmt oder unsicher sein. In dieser Konstellation besteht eine hohe Notwendigkeit von Zusatzdiagnostik, um neurologische Erkrankungen auszuschließen, die ebenfalls Muskelschwäche (Paresen), Muskelschwund (Myatrophien) oder Muskelsteifigkeit (Spastik) verursachen können. Wichtige neurologische „Tests“ sind die Magnetresonanztomographie (MRT) des Gehirns (zerebrale MRT) und des Rückenmarks (spinale MRT). Weitere Zusatzdiagnostik kann die die Elektromyographie (EMG), Elektroneurographie sowie Labordiagnostik einschließlich Biomarker in Blut und Nervenwasser (Liquor cerebrospinalis) umfassen.
-
Gibt es Regionen mit einer besonderen ALS-Häufigkeit?
In Deutschland und Europa ist von einer ähnlichen Häufigkeit auszugehen. Dennoch sind regionale Unterschiede möglich, deren Ursachen noch nicht geklärt sind. Wiederholt wurden kleinere Orte beschrieben, in denen mehrere Menschen an ALS zeitgleich erkrankt sind. Diese Konstellationen waren über Jahrzehnte der Anlass für Hypothesen von Umweltfaktoren, die eine regionale oder kollektive Häufung von ALS bedingen. Diese externen Faktoren konnten bisher nicht identifiziert werden. Auch ist bisher unklar, ob es sich bei den Berichten tatsächlich um regionale Häufungen oder vielmehr um statistische Phänomene handelt. Darunter ist zu verstehen, dass im Auftreten von Erkrankungen keine statistische „Gleichverteilung“ besteht. Dieses Phänomen ist beim Würfelspiel zu beobachten – so kann eine Zahl für mehrere Durchgänge besonderes häufig oder sogar hintereinander gewürfelt wird, obwohl die Zahl auf dem Würfel nur einmal vorkommt. In gleicher Weise ist erklärbar, dass die die Diagnose einer ALS mehrfach hintereinander in der gleichen Gruppe gestellt wird. Im Gegensatz zu statistischen Effekten (einer scheinbaren ALS-Häufung) ist zu vermuten, dass genetische Faktoren zu einer realen Häufung der ALS in bestimmten Regionen beitragen. So kann es in Regionen mit geringer genetischen „Durchmischung“ (z. B. durch Abgeschiedenheit der Bevölkerung auf Inseln) zu einem erhöhten genetischen Risiko der ALS kommen.
-
Ist die ALS schwer zu diagnostizieren?
Die ALS ist eine schwere Erkrankung, die jedoch relativ leicht zu diagnostizieren ist. Für Fachärztinnen oder Fachärzte für Neurologie sind die Symptome und diagnostischen Kriterien sehr geläufig. Bereits durch die äußeren Symptome (Muskelschwund, Schwäche, Reflexveränderungen, veränderte Muskelspannung bis hin zur Spastik, Faszikulationen, verminderte Beweglichkeit der Zunge, Gewichtsabnahme etc.) entstehen Verdachtsmomente einer ALS. Die einzelnen Symptome für sich genommen sind nicht beweisend. Erst eine typische Kombination der klinischen Merkmale (z. B. die Kombination von Muskelschwund mit einer Schwäche und Reflexsteigerung) sind charakteristisch für die ALS-Diagnose. Bereits durch die „einfache“ körperliche Untersuchung lässt sich die Verdachtsdiagnose einer ALS formulieren. In einem zweiten Schritt folgen medizintechnische, laborchemische und radiologische Untersuchungen, mit denen andere neurologische Erkrankungen (die im Einzelfall ähnliche Krankheitssymptome verursachen können) ausgeschlossen werden. In einer neurologischen Schwerpunktpraxis oder während eines Krankenhausaufenthaltes werden die technischen Untersuchungen durchgeführt (Nadel-Untersuchung der Muskulatur, Elektrische Messung der Nerven, Analyse des Nervenwassers, Magnetresonanztomographie). Nach Abschluss der technischen Zusatzuntersuchungen lässt sich in der Mehrheit der Betroffenen die ALS-Diagnose ohne Zweifel feststellen. Lediglich bei einer Minderheit der ALS-Patientinnen und ‑Patienten ist die Diagnosestellung kompliziert und mit weiteren Untersuchungsschritten verbunden (z. B. Nerven- und Muskelbiopsie). Bei der Mehrheit der Betroffenen lässt sich bereits mit wenigen Untersuchungsschritten (körperlich-neurologische Untersuchung und Elektromyographie) die Diagnose einer ALS formulieren. Einen Teil der Menschen, die eine ALS-Diagnose erhalten, reagiert mit Skepsis und Zweifeln, dass eine so schwerwiegende Diagnose mit „einfachsten“ Mitteln („nur mit körperlichen Übungen und Reflexhammer“, ohne „richtige“ Diagnoseverfahren) zu diagnostizieren ist. Insgesamt liegt die Herausforderung in der Diagnosestellung der ALS nicht in den apparativ-technischen Aufwendungen, sondern vielmehr in den Erläuterungen und der Verarbeitung der mitgeteilten Diagnose.
-
Ist es sinnvoll, eine „zweite Meinung“ zur Diagnose und Prognosestellung der ALS einzuholen?
Die Ermittlung einer zweiten Meinung ist ein etabliertes Konzept bei schweren, komplexen und seltenen Erkrankungen. Das Grundprinzip besteht darin, die diagnostische und prognostische Einschätzung der ALS durch eine andere Neurologin oder einen anderen Neurologen mit Spezialisierung und Erfahrung in der Diagnose und Behandlung der ALS einzuholen. Der Vorteil einer zweiten Meinung liegt darin, dass verschiedene Ärzte über unterschiedliche Erfahrungs- und Behandlungsschwerpunkte verfügen. Auch die Herangehensweise und Kommunikation der offenen Fragen kann sehr unterschiedlich sein. Diese Nuancen in der Beratung und Einschätzung können für die Betroffenen von Interesse sein. Die zweite Meinung zielt nicht darauf ab, eine „bessere“ Meinung zu erhalten, sondern eine zusätzliche und ergänzende Einschätzung zu erfahren. Nur im Ausnahmefall führt die Einholung einer zweiten Meinung zu einer grundsätzlich anderen diagnostischen und prognostischen Einschätzung. Wahrscheinlicher ist, dass in der zweiten Meinung das Konzept der Erstbehandlung bestätigt wird. Selbst in dieser Situation ist von einer Stärkung der Arzt-Patientenbeziehung zum erstbehandelnden Arzt auszugehen. Nachteile im Einholen der zweiten Meinung liegen in den Aufwendungen und Belastungen, die mit überregionalen Anreisen und Transporten verbunden seien können. Daher ist auch der Verzicht auf eine zweite Meinung gleichberechtigt und nachvollziehbar. Auch die Beschränkung von Informationen und der bewusste Verzicht auf eine allumfassende Beratung (einschließlich einer zweiten Meinung) liegt im Ermessen der individuellen Patientinnen und Patienten. Daher ist die zweite Meinung eine grundsätzliche Möglichkeit der medizinischen Beratung bei der ALS, die jedoch noch nicht für alle zur Anwendung kommt.
-
Ist eine Vorhersage des Verlaufes möglich?
Der Verlauf der ALS ist sehr unterschiedlich und individuell. Die Entwicklung der Symptome wird von der konkreten Verlaufsform der ALS bestimmt. Eine grundsätzliche Unterscheidung zwischen einem schnellen, mittleren oder langsamen Krankheitsverlauf ist möglich. Dabei wird berücksichtigt, mit welcher Dynamik die motorischen Symptome fortschreiten. Von besonderer Bedeutung ist die Ausbreitung von Lähmungen (und damit verbundenen motorischen Defiziten) von einer betroffenen Körperregion auf eine nachfolgende Region (Ausbreitungsdynamik). Die Entwicklung der motorischen Defizite wird anhand der ALS-Funktionsskala (englisch: ALS Functional Rating Scale, ALS-FRS) ermittelt. Weitere Verlaufskriterien sind die Entwicklung des Körpergewichtes (Body-Maß-Index), der Atemfunktionen und des Biomarkers Neurofilament Light Chain (NF‑L). Eine Vorhersage des ALS-Verlaufes setzt eine Berücksichtigung, Interpretation und Abwägung einzelner Faktoren und ihrer Kombination voraus. Die Einschätzung des individuellen Verlaufes erfolgt durch Neurologinnen und Neurologen, die sich zur Diagnose und Behandlung der ALS spezialisiert haben.
-
Verläuft die ALS in Schüben?
Die ALS verläuft zumeist in langsam fortschreitender Weise. In seltenen Konstellationen kann der Erkrankungsbeginn im Verlauf weniger Stunden oder Tage beginnen, so dass der frühe Verlauf im Sinne eines „Schubes“ oder einer Akutsymptomatik erscheint. Weiterhin ist möglich, dass Patientinnen und Patienten bestimmte Funktionen von einem zum anderen Tag nicht mehr realisieren können. Dieses Funktionsdefizit kann ebenfalls als „Schub“ erlebt werden, obwohl der zugrundeliegende Prozess der Neurodegeneration weitgehend kontinuierlich ist. Dieses Phänomen ist im Sinne des sprichwörtlichen Tropfens zu verstehen, der das Glas (der Erkrankung) zum Überlaufen bringt.